[1]:
import pandas as pd
import networkx as nx
from gsnn.simulate.nx2pyg import nx2pyg
from gsnn.models.GSNN import GSNN
from gsnn.models.NN import NN
import numpy as np
import torch
from scipy.stats import spearmanr
from rdkit import Chem
from rdkit.Chem import rdFingerprintGenerator
from sklearn.metrics import r2_score
from matplotlib import pyplot as plt
from gsnn.interpret.GSNNExplainer import GSNNExplainer
from gsnn.interpret.ContrastiveIGExplainer import ContrastiveIGExplainer
from gsnn.interpret.CounterfactualExplainer import CounterfactualExplainer
from gsnn.interpret.NoiseTunnel import NoiseTunnel
import umap
from sklearn.decomposition import PCA
import seaborn as sbn
torch.manual_seed(0)
np.random.seed(0)
%load_ext autoreload
%autoreload 2
/home/teddy/miniconda3/envs/gsnn-mds/lib/python3.12/site-packages/tqdm/auto.py:21: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html
from .autonotebook import tqdm as notebook_tqdm
DrugCell implementation example
In this tutorial, we will implement the drugcell model using a GSNN and train using their provided data.
Predicting Drug Response and Synergy Using a Deep Learning Model of Human Cancer Cells.Kuenzi, Brent M. et al. Cancer Cell, Volume 38, Issue 5, 672 - 684.e6
The code below assumes you have cloned the drugcell repo, and the drugcell_path is set correctly below.
[2]:
drugcell_path = '/home/teddy/local/DrugCell/'
[3]:
cell2ind = pd.read_csv(f'{drugcell_path}/data/cell2ind.txt', sep='\t', header=None, index_col=0)
cell2mut = pd.read_csv(f'{drugcell_path}/data/cell2mutation.txt', sep=',', header=None)
drug2ind = pd.read_csv(f'{drugcell_path}/data/drug2ind.txt', sep='\t', header=None, index_col=0)
drugcell_ont = pd.read_csv(f'{drugcell_path}/data/drugcell_ont.txt', sep='\t', header=None).rename(columns={0:'target', 1:'source', 2:'edge_type'})
gene2ind = pd.read_csv(f'{drugcell_path}/data/gene2ind.txt', sep='\t', header=None, index_col=0)
drugcell_train = pd.read_csv(f'{drugcell_path}/data/drugcell_train.txt', sep='\t', header=None)
drugcell_test = pd.read_csv(f'{drugcell_path}/data/drugcell_test.txt', sep='\t', header=None)
drugcell_val = pd.read_csv(f'{drugcell_path}/data/drugcell_val.txt', sep='\t', header=None)
[4]:
# zscore y
y_mu = drugcell_train[2].mean()
y_std = drugcell_train[2].std()
drugcell_train[2] = (drugcell_train[2] - y_mu) / y_std
drugcell_test[2] = (drugcell_test[2] - y_mu) / y_std
drugcell_val[2] = (drugcell_val[2] - y_mu) / y_std
[5]:
input_nodes = gene2ind[1].values.tolist()
function_nodes = list(set(drugcell_ont[lambda x: x.edge_type == 'default'].source.tolist()).union(set(drugcell_ont[lambda x: x.edge_type == 'default'].target.tolist())))
output_nodes = [f'OUT{i}__GO:0008150' for i in range(6)] # 32 cell dim
print(f'{len(input_nodes)} input nodes')
print(f'{len(function_nodes)} function nodes')
print(f'{len(output_nodes)} output nodes')
3008 input nodes
2086 function nodes
6 output nodes
[6]:
G = nx.DiGraph()
for i, row in drugcell_ont.iterrows():
G.add_edge(row['source'], row['target'], edge_type='input')
# add multiple output nodes to create multidimensional mutation representation
for out in output_nodes:
G.add_edge('GO:0008150', out)
print(f'# nodes: {len(G)}')
print(f'# edges: {len(G.edges())}')
# nodes: 5100
# edges: 62926
[7]:
data = nx2pyg(G, input_nodes, function_nodes, output_nodes)
[8]:
class DrugCellDataset(torch.utils.data.Dataset):
def __init__(self, data, cell2code, drug2code):
self.data = data.reset_index(drop=True)
self.cell2code = cell2code
self.drug2code = drug2code
def __len__(self):
return len(self.data)
def __getitem__(self, idx):
obs = self.data.iloc[idx]
x_cell = torch.tensor(self.cell2code[obs[0]], dtype=torch.float32)
x_drug = torch.tensor(self.drug2code[obs[1]], dtype=torch.float32)
y = torch.tensor(obs[2], dtype=torch.float32)
return x_cell, x_drug, y
[9]:
drug2code = {}
smiles = np.unique( drugcell_train[1].tolist() + drugcell_test[1].tolist() + drugcell_val[1].tolist())
mfpgen = rdFingerprintGenerator.GetMorganGenerator(radius=2, fpSize=2048)
for i, s in enumerate(smiles):
print(f'{i}/{len(smiles)}', end='\r')
mol = Chem.MolFromSmiles(s)
fp = mfpgen.GetFingerprint(mol)
code = np.array(fp.ToList(), dtype=np.float32)
drug2code[s] = code
cell2code = {}
cell_muts = cell2mut.merge(cell2ind.rename(columns={1:'cell_id'}), left_index=True, right_index=True)
for i, row in cell_muts.iterrows():
print(f'{i}/{len(cell2mut)}', end='\r')
cell2code[row['cell_id']] = np.array(row.values[:-1], dtype=np.float32)
1224/1225
[10]:
train_dataset = DrugCellDataset(drugcell_train, cell2code, drug2code)
test_dataset = DrugCellDataset(drugcell_test, cell2code, drug2code)
val_dataset = DrugCellDataset(drugcell_val, cell2code, drug2code)
train_loader = torch.utils.data.DataLoader(train_dataset, batch_size=128, shuffle=True, num_workers=4)
test_loader = torch.utils.data.DataLoader(test_dataset, batch_size=512, shuffle=False)
val_loader = torch.utils.data.DataLoader(val_dataset, batch_size=512, shuffle=False)
print(f'{len(train_dataset)} train samples')
print(f'{len(test_dataset)} test samples')
print(f'{len(val_dataset)} val samples')
print(f'{len(train_dataset) + len(test_dataset) + len(val_dataset)} total samples')
10000 train samples
1000 test samples
1000 val samples
12000 total samples
[11]:
class DrugCell(torch.nn.Module):
def __init__(self, gsnn_kwargs, nn_kwargs):
super().__init__()
self.cell_encoder = GSNN(**gsnn_kwargs)
self.drug_encoder = NN(**nn_kwargs)
self.nn = torch.nn.Sequential(
torch.nn.LazyLinear(6),
torch.nn.BatchNorm1d(6),
torch.nn.ELU(),
torch.nn.Linear(6,1))
def forward(self, x_cell, x_drug, edge_mask=None):
x_cell = self.cell_encoder(x_cell, edge_mask=edge_mask)
x_drug = self.drug_encoder(x_drug)
x = torch.cat([x_cell, x_drug], dim=1)
x = self.nn(x)
return x
[12]:
gsnn_kwargs = {'edge_index_dict':data.edge_index_dict,
'node_names_dict':data.node_names_dict,
'channels':5,
'layers':7,
'dropout':0.2,
'share_layers':False,
'add_function_self_edges':True,
'norm':'layer',
'norm_first':True,
'init':'degree_normalized',
'node_attn':True,
'bias':True,
'checkpoint':True,
'residual':True,
'node_mlp_hidden':256}
nn_kwargs = {'in_channels':2048,
'hidden_channels':100,
'out_channels':6,
'layers':2,
'dropout':0.2,
'nonlin':torch.nn.ELU,
'out':None,
'norm':torch.nn.BatchNorm1d}
device = torch.device('cuda' if torch.cuda.is_available() else 'cpu')
model = DrugCell(gsnn_kwargs, nn_kwargs).to(device)
print('# params [gsnn; cell encoder]', sum([p.numel() for p in model.cell_encoder.parameters()]))
print('# params [nn; drug encoder]', sum([p.numel() for p in model.drug_encoder.parameters()]))
optim = torch.optim.AdamW(model.parameters(), lr=1e-3, weight_decay=1e-2)
crit = torch.nn.MSELoss()
# params [gsnn; cell encoder] 3047982
# params [nn; drug encoder] 216006
[13]:
best_loss = np.inf
best_state_dict = None
for epoch in range(10):
torch.cuda.empty_cache()
model.train()
train_loss = 0
for i, (x_cell, x_drug, y) in enumerate(train_loader):
optim.zero_grad()
yhat = model(x_cell.to(device), x_drug.to(device))
loss = crit(yhat.squeeze(), y.to(device))
loss.backward()
optim.step()
print(f'[batch {i}/{len(train_loader)}, loss: {loss:.4f}]', end='\r')
train_loss += loss.item()
torch.cuda.empty_cache()
model.eval()
val_loss = 0 ; val_r2 = 0 ; val_sr = 0
with torch.no_grad():
for x_cell, x_drug, y in val_loader:
yhat = model(x_cell.to(device), x_drug.to(device))
loss = crit(yhat.squeeze(), y.to(device))
val_loss += loss.item()
val_r2 += r2_score(y.cpu().numpy().ravel(), yhat.cpu().numpy().ravel())
val_sr += spearmanr(y.cpu().numpy().ravel(), yhat.cpu().numpy().ravel())[0]
train_loss /= len(train_loader)
val_loss /= len(val_loader)
val_r2 /= len(val_loader)
val_sr /= len(val_loader)
# update and detach model state dict
if val_loss < best_loss:
best_loss = val_loss
best_state_dict = {k:v.clone().detach().cpu() for k, v in model.state_dict().items()}
print(f'epoch: {epoch}, train_loss: {train_loss:.4f}, val_loss: {val_loss:.4f}, val_r2: {val_r2:.4f}, val_sr: {val_sr:.4f}')
epoch: 0, train_loss: 0.6825, val_loss: 0.5607, val_r2: 0.4683, val_sr: 0.6584
epoch: 1, train_loss: 0.5412, val_loss: 0.5270, val_r2: 0.5007, val_sr: 0.6913
epoch: 2, train_loss: 0.5144, val_loss: 0.4988, val_r2: 0.5283, val_sr: 0.7043
epoch: 3, train_loss: 0.4880, val_loss: 0.4865, val_r2: 0.5404, val_sr: 0.7147
epoch: 4, train_loss: 0.4625, val_loss: 0.4836, val_r2: 0.5442, val_sr: 0.7106
epoch: 5, train_loss: 0.4322, val_loss: 0.4794, val_r2: 0.5472, val_sr: 0.7190
epoch: 6, train_loss: 0.4188, val_loss: 0.4784, val_r2: 0.5494, val_sr: 0.7182
epoch: 7, train_loss: 0.4043, val_loss: 0.4864, val_r2: 0.5411, val_sr: 0.7145
epoch: 8, train_loss: 0.3943, val_loss: 0.4825, val_r2: 0.5453, val_sr: 0.7199
epoch: 9, train_loss: 0.3843, val_loss: 0.4658, val_r2: 0.5604, val_sr: 0.7229
[14]:
# authors of drugcell report avg spearman corr of 0.8 (5-fold cross-validation) on the full 509,294 observation dataset (Here we use the provided 12k observation subset)
# (https://www.cell.com/cancer-cell/fulltext/S1535-6108(20)30488-8?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS1535610820304888%3Fshowall%3Dtrue#sec-4)
model.load_state_dict(best_state_dict)
model.to(device)
model.eval()
ys = []
yhats = []
with torch.no_grad():
for x_cell, x_drug, y in test_loader:
yhats.append(model(x_cell.to(device), x_drug.to(device)).detach().cpu().numpy() )
ys.append(y.detach().cpu().numpy().ravel())
y = np.concatenate(ys)
yhat = np.concatenate(yhats)
# unscale to get back to AUC
y = y*y_std + y_mu
yhat = yhat*y_std + y_mu
mse = np.mean((y - yhat)**2)
r2 = r2_score(y, yhat)
sr = spearmanr(y, yhat)
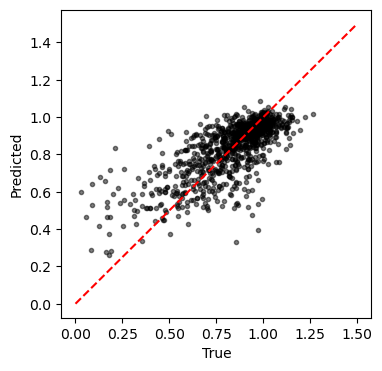
print('Test results:')
print(f'\tMSE: {mse:.4f}')
print(f'\tR2: {r2:.4f}')
print(f'\tSpearman: {sr[0]:.4f}')
Test results:
MSE: 0.0626
R2: 0.5372
Spearman: 0.7201
[15]:
plt.figure(figsize=(4,4))
plt.plot(y, yhat, 'k.', alpha=0.5)
plt.plot([0, 1.5], [0, 1.5], 'r--')
plt.xlabel('True')
plt.ylabel('Predicted')
plt.show()
GSNN Explainer to investigate drug response
[16]:
# I don't have access to drug names, just the smiles, so we're going to arbitrarily pick one that has a good number of obs
smile = list(drug2code.keys())[200]
print('drug smiles to explain sensitivity:', smile)
subset = drugcell_train[lambda x: x[1] == smile]
print('# obs', subset.shape[0])
drug_dataset = DrugCellDataset(subset, cell2code, drug2code)
drug_loader = torch.utils.data.DataLoader(drug_dataset, batch_size=100, shuffle=False)
x_cell, x_drug, y = next(iter(drug_loader))
yhat = model(x_cell.to(device), x_drug.to(device))
drug smiles to explain sensitivity: CC(C)(C)OC(=O)NC1=CC=C(C=C1)C2=CC(=NO2)C(=O)NCCCCCCC(=O)NO
# obs 37
[17]:
# first, we'll find some cell latent features that are correlated with the drug sensitivity
gsnn = model.cell_encoder
z_cell = gsnn(x_cell.to(device)).detach().cpu().numpy()
zy_corr_res = {'ix':[], 'corr':[], 'pval':[]}
for i in range(z_cell.shape[1]):
zc = z_cell[:, i]
corr, pval = spearmanr(zc, y)
zy_corr_res['ix'].append(i)
zy_corr_res['corr'].append(corr)
zy_corr_res['pval'].append(pval)
zy_corr_res = pd.DataFrame(zy_corr_res).sort_values('pval', ascending=True)
zy_corr_res.head()
[17]:
| ix | corr | pval | |
|---|---|---|---|
| 5 | 5 | 0.644381 | 0.000017 |
| 4 | 4 | 0.642722 | 0.000018 |
| 0 | 0 | -0.639640 | 0.000020 |
| 1 | 1 | -0.634187 | 0.000025 |
| 3 | 3 | 0.393789 | 0.015889 |
[18]:
# use GSNNExplainer to identify the edges that are important for predicting a given target (cell representation)
torch.cuda.empty_cache()
explainer = GSNNExplainer(gsnn, data, ignore_cuda=False, gumbel_softmax=True, hard=True, tau0=10, min_tau=0.5,
prior=5, iters=2000, lr=1e-2, weight_decay=0, entropy=10,
beta=1e-3, verbose=True, optimizer=torch.optim.Adam, free_edges=0)
res = explainer.explain(x_cell.to(device), targets=[zy_corr_res.ix.values[0]])
res.sort_values('score', ascending=False)
iter: 1999 | loss: 0.1916 | mse: 0.0560 | r2: 0.929 | active edges: 172 / 65012 | entropy: 0.0036711
==================================================
POST-TRAINING EVALUATION (edges > 0.5)
==================================================
Selected edges: 144 / 65012 (0.2%)
MSE (subset): 0.050052
R² (subset): 0.9365
Variance explained: 0.9413
Correlation: 0.9702
==================================================
[18]:
| source | target | score | |
|---|---|---|---|
| 2287 | GO:0007399 | GO:0008150 | 0.999982 |
| 1050 | GO:0010605 | GO:0008150 | 0.999958 |
| 2406 | GO:0051606 | GO:0008150 | 0.999883 |
| 3154 | GO:0071243 | GO:0008150 | 0.999728 |
| 3103 | GO:0032970 | GO:0008150 | 0.999708 |
| ... | ... | ... | ... |
| 38720 | GHRL | GO:0016525 | 0.000026 |
| 1094 | GO:0031057 | GO:0033044 | 0.000025 |
| 56596 | PCSK9 | GO:0042632 | 0.000025 |
| 13295 | ATP1A1 | GO:0086002 | 0.000024 |
| 47948 | RXRB | GO:0006367 | 0.000023 |
65012 rows × 3 columns
[19]:
plt.figure()
plt.hist(res.score)
plt.yscale('log')
plt.xlabel('edge score')
plt.show()
[20]:
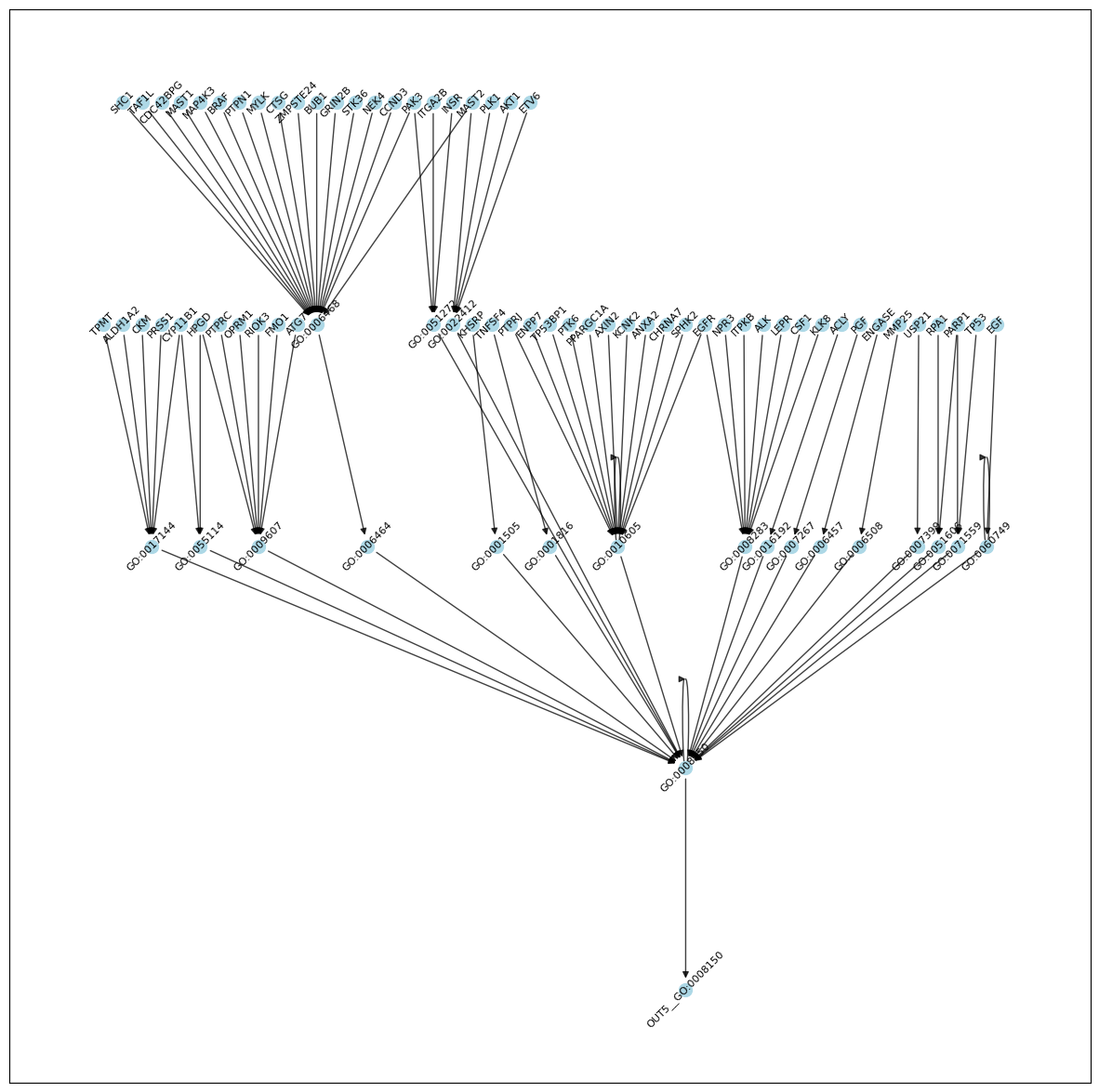
print('# edges with score > 0.5:', res[lambda x: x.score > 0.5].shape[0])
# edges with score > 0.5: 144
[21]:
G = nx.from_pandas_edgelist(res[lambda x: x.score > 0.5], source='source', target='target', create_using=nx.DiGraph)
# remove nodes that are not a descendent of a gene node (leftovers from thresholding)
# or are not a ancestor of the output node
n = set()
for g in input_nodes:
if g in G:
n.update(nx.descendants(G, g))
n.update(set([g]))
n = n.intersection(set(nx.ancestors(G, output_nodes[zy_corr_res.ix.values[0]])).union(set([output_nodes[zy_corr_res.ix.values[0]]])))
G = G.subgraph(n).copy()
# use heiarchichal "dot" layout
H = nx.convert_node_labels_to_integers(G, label_attribute="node_label")
H_layout = nx.nx_pydot.pydot_layout(H, prog="dot")
pos = {H.nodes[n]["node_label"]: p for n, p in H_layout.items()}
plt.figure(figsize=(15,15))
nx.draw_networkx_edges(G, pos, width=1.0, alpha=0.75)
nx.draw_networkx_nodes(G, pos, node_size=100, node_color='lightblue')
nl = nx.draw_networkx_labels(G, pos, font_size=8, )
for _, label in nl.items():
label.set_rotation(45) # Rotate labels by 45 degrees
plt.show()
Contrastive Explanation of drug sensitivity
For a given drug we will choose two samples, a sensitive and a resistant, and try to understand which edges can be attributed to the difference in response.
Note that we are explaining predicted AUC in this example, not just a single gsnn output target (as was the case above).
[22]:
ix_res = yhat.argmax()
ix_sens = yhat.argmin()
yhat_res = model(x_cell[ix_res].cuda().view(1,-1), x_drug[ix_res].cuda().view(1,-1))
yhat_sens = model(x_cell[ix_sens].cuda().view(1,-1), x_drug[ix_sens].cuda().view(1,-1))
print(f'yhat_res: {yhat_res.item():.4f} [true: {y[ix_res]:.4f}]')
print(f'yhat_sens: {yhat_sens.item():.4f} [true: {y[ix_sens]:.4f}]')
x1_cell = x_cell[ix_res]
x1_drug = x_drug[ix_res]
x2_cell = x_cell[ix_sens]
x2_drug = x_drug[ix_sens]
x1 = torch.cat([x1_cell, x1_drug], dim=0)
x2 = torch.cat([x2_cell, x2_drug], dim=0)
class wrapper(torch.nn.Module):
def __init__(self, model, x_cell_len):
super().__init__()
self.model = model
self.edge_index = model.cell_encoder.edge_index
self.homo_names = model.cell_encoder.homo_names
self.x_cell_len = x_cell_len
def __call__(self, x, edge_mask=None):
x_cell = x[:, :self.x_cell_len]
x_drug = x[:, self.x_cell_len:]
return self.model(x_cell, x_drug, edge_mask=edge_mask)
yhat_res: -0.7116 [true: 0.9106]
yhat_sens: -0.7250 [true: -1.7946]
[23]:
explainer = NoiseTunnel(ContrastiveIGExplainer(
wrapper(model.eval(), x_cell.shape[1]), data, n_steps=50),
n_samples=50, noise_std=0.1)
ig_res = explainer.explain(x1, x2, target_idx=0)
ig_res = ig_res.sort_values('score', ascending=False)
ig_res.head(10)
[23]:
| source | target | score | |
|---|---|---|---|
| 2939 | GO:0045937 | GO:0008150 | 0.018616 |
| 1000 | GO:0008283 | GO:0008150 | 0.011895 |
| 62921 | GO:0008150 | OUT1__GO:0008150 | 0.011048 |
| 2287 | GO:0007399 | GO:0008150 | 0.010690 |
| 1796 | GO:0051302 | GO:0008150 | 0.010184 |
| 2409 | GO:0042752 | GO:0008150 | 0.009623 |
| 548 | GO:0006464 | GO:0008150 | 0.009523 |
| 3017 | GO:0002504 | GO:0008150 | 0.009221 |
| 2125 | GO:0016192 | GO:0008150 | 0.007903 |
| 1509 | GO:0010557 | GO:0008150 | 0.007854 |
[24]:
# For the mutations that are most implicated to explain the difference in drug sensitivity, let's confirm that the mutations are indeed different
for g in ig_res[lambda x: x.source.isin(input_nodes)].head(10).source.values:
mut_ix = gene2ind[1].values.tolist().index(g)
print(f'Gene: {g} >> x1_cell[mut_ix]: {x1_cell[mut_ix]}, x2_cell[mut_ix]: {x2_cell[mut_ix]}')
Gene: HSP90AA1 >> x1_cell[mut_ix]: 0.0, x2_cell[mut_ix]: 1.0
Gene: PARP14 >> x1_cell[mut_ix]: 0.0, x2_cell[mut_ix]: 1.0
Gene: PRKD1 >> x1_cell[mut_ix]: 0.0, x2_cell[mut_ix]: 1.0
Gene: KLK8 >> x1_cell[mut_ix]: 0.0, x2_cell[mut_ix]: 1.0
Gene: FMO1 >> x1_cell[mut_ix]: 0.0, x2_cell[mut_ix]: 1.0
Gene: CASP8 >> x1_cell[mut_ix]: 0.0, x2_cell[mut_ix]: 1.0
Gene: EPX >> x1_cell[mut_ix]: 0.0, x2_cell[mut_ix]: 1.0
Gene: ADAM15 >> x1_cell[mut_ix]: 0.0, x2_cell[mut_ix]: 1.0
Gene: ITGB7 >> x1_cell[mut_ix]: 0.0, x2_cell[mut_ix]: 1.0
Gene: LTK >> x1_cell[mut_ix]: 0.0, x2_cell[mut_ix]: 1.0
[25]:
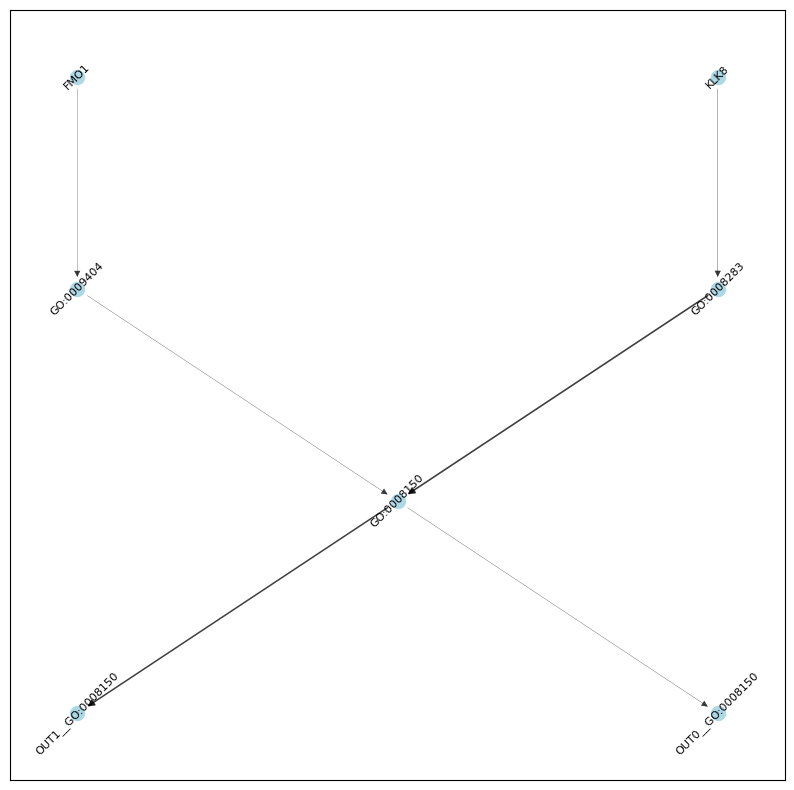
G = nx.from_pandas_edgelist(ig_res[lambda x: x.score > 0.002], source='source', target='target', edge_attr='score', create_using=nx.DiGraph)
# TO make the graph more readable...
# subset to only nodes that are ancestors or descendants of GO:0008150 (biological_process) AND descendent of a gene node
n = nx.ancestors(G, 'GO:0008150').union(nx.descendants(G, 'GO:0008150')).union(set(['GO:0008150']))
n2 = set()
for g in input_nodes:
if g in G:
n2.update(nx.descendants(G, g))
n2.update(set([g]))
n = n.intersection(n2)
G = G.subgraph(n).copy()
# use heiarchichal "dot" layout
H = nx.convert_node_labels_to_integers(G, label_attribute="node_label")
H_layout = nx.nx_pydot.pydot_layout(H, prog="dot")
pos = {H.nodes[n]["node_label"]: p for n, p in H_layout.items()}
edge_widths = [G.edges[e]['score'] * 100 for e in G.edges]
plt.figure(figsize=(10,10))
nx.draw_networkx_edges(G, pos, alpha=0.75, width=edge_widths)
nx.draw_networkx_nodes(G, pos, node_size=100, node_color='lightblue')
nl = nx.draw_networkx_labels(G, pos, font_size=8, )
for _, label in nl.items():
label.set_rotation(45) # Rotate labels by 45 degrees
plt.show()
Node activations
[26]:
torch.cuda.empty_cache()
x_cell, x_drug, y = next(iter(test_loader))
with torch.no_grad():
node_acts = model.cell_encoder.get_node_activations(x_cell.cuda(), agg='sum')
[27]:
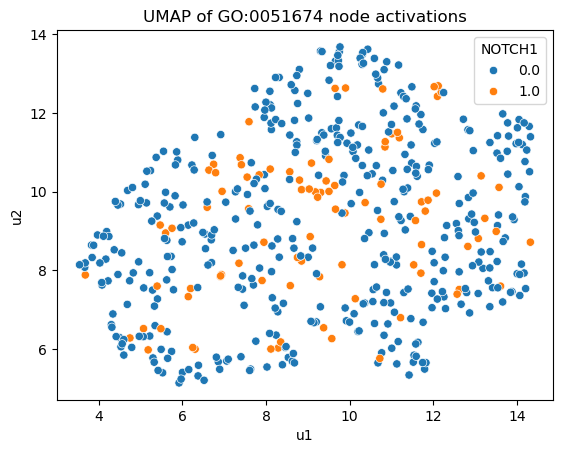
reducer = umap.UMAP(n_components=2, min_dist=0.5, n_neighbors=25)
u = reducer.fit_transform(node_acts['GO:0006955'].detach().cpu().numpy())
df = pd.DataFrame(u, columns=['u1', 'u2'])
for i,row in gene2ind.iterrows():
df = df.assign(**{row[1]: x_cell[:,i]})
plt.figure()
sbn.scatterplot(x='u1', y='u2', hue='NOTCH1', data=df)
plt.title('UMAP of GO:0051674 node activations')
plt.show()